在蛋白互作研究中,传统方法如酵母双杂交(Y2H)与免疫沉淀-质谱联用(IP-MS)面临诸多限制;
1、实验流程繁琐: Y2H需构建文库并进行多轮筛选,假阳性率高(>30%)
2、周期较长: IP-MS从样本制备至数据分析通常需 2–4 周
3、识别盲区: 难以检测瞬时或弱亲和互作
我们基于AlphaFold3构建多模态AI互作预测平台,实现从序列到结构预测的智能化、高通量蛋白互作筛选。
| 维度 | 传统方法 | 致一平台优势 |
| 筛选周期 | 周-月级 | <24小时(预测 + 验证) |
| 实验成本 | 单次实验5–10K元 | 大幅下降(纯计算流程) |
| 数据维度 | 二元互作 | 支持多蛋白复合体预测 |
| 模块 | 技术特点 | 功能描述 |
| MegaDock | 快速FFT算法,召回率>95% | 海量初筛互作对 |
| Hdock | 柔性对接 + 多目标遗传算法> | 精筛高亲和力互作模型 |
| AlphaFold3 | 等变注意力神经网络 | 验证复合物结构可信度 |
1、构建蛋白结构数据库(AF3建模)
2、诱饵蛋白结构准备
3、Hdock优化筛选TOP10
4、AlphaFold3精建复合物结构,计算iPTM/PTM值

| 项目内容 | 技术特点 |
| 候选互作蛋白列表(TOP10) | 包含UniProt编号、结合能、预测结构链接 |
| 精筛结构模型(.pdb) | 诱饵与互作蛋白复合物结构文件 |
| iPTM/PTM值表格 | 验证互作稳定性与建模可信度 |
| 对接界面分析图 | 显示结合关键残基、氢键、疏水作用图谱 |
| 交互报告与筛选流程说明文档 | 附带筛选参数、分析策略与后续验证建议 |
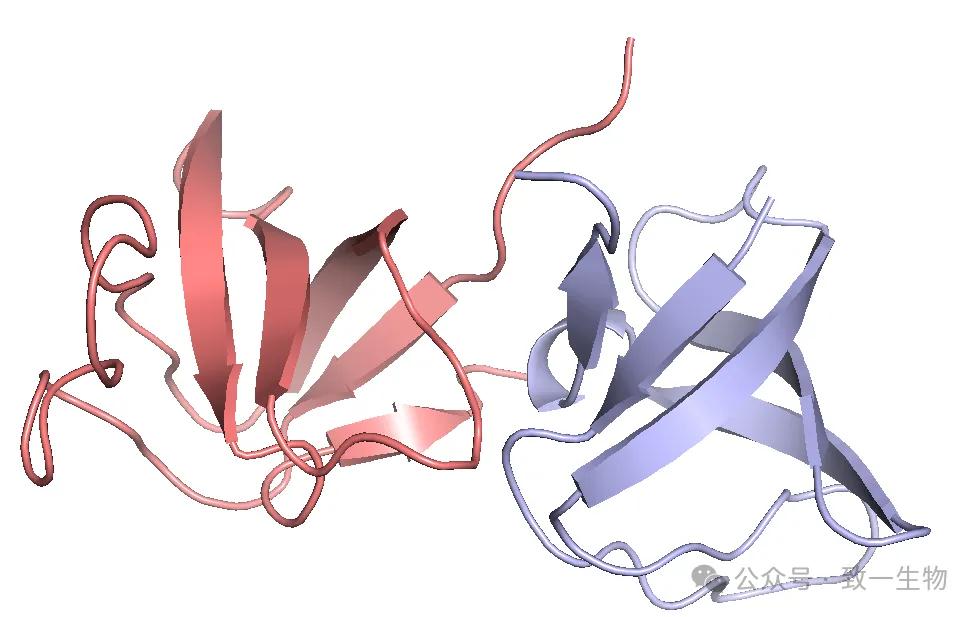
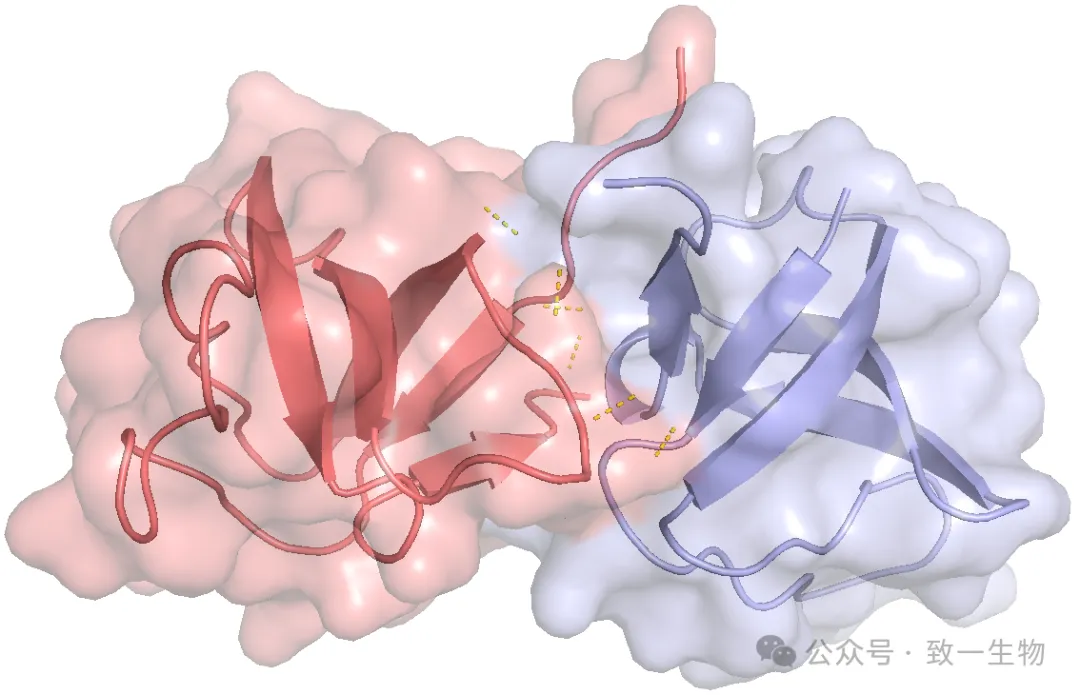
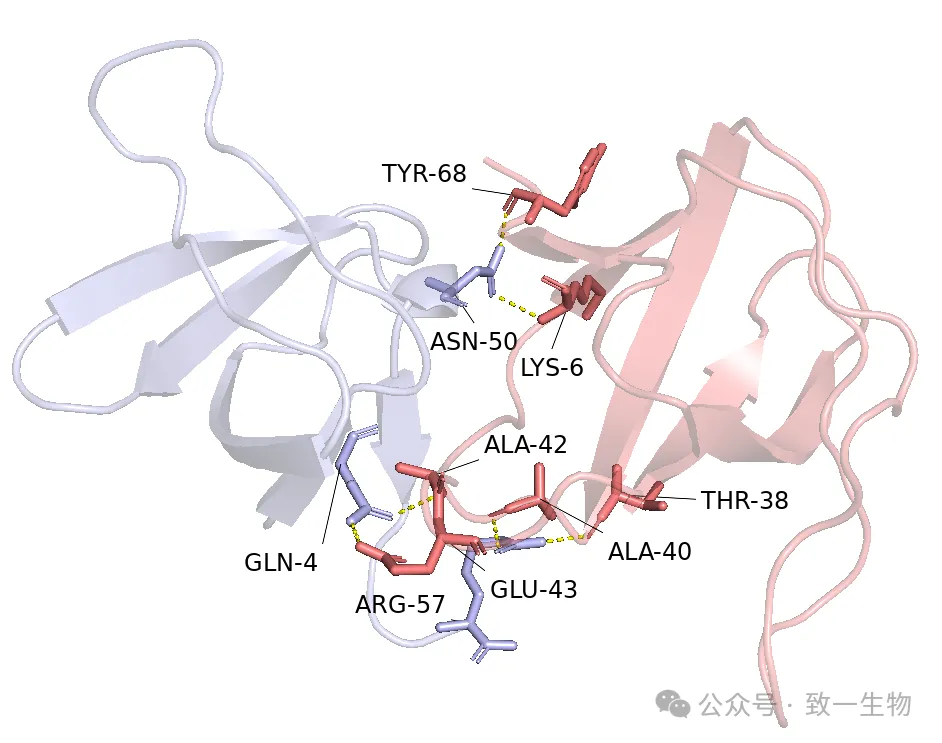
[1] Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
[2] Yan, Y. et al. HDOCK: a web server for protein–protein and protein–DNA/RNA docking. Nucleic Acids Res. 45(W1), W365–W373 (2017).